Blog Categories
ScaleSC: Supercharging Single Cell RNA-seq Analysis with GPU Power
Single cell RNA sequencing(scRNA-seq) has transformed our understanding of cellular biology by allowing us to examine gene expression in individual cells. However, this powerful technology comes with a significant computational cost. In a revolutionary development for the single cell genomics community, Zhengyu Ouyang at BioInfoRx, along with researchers from Biogen, has introduced ScaleSC, a GPU-accelerated solution for single cell data processing (Hu et al., 2025).
Existing popular tools such as Scanpy (Wolf et al., 2018) and rapids-singlecell (Dicks et al., 2024) facilitate single cell data analysis, but they still have notable limitations. Scanpy relies on CPU-based matrix calculations, whereas rapids-singlecell is a GPU-accelerated scRNA-seq analysis package, requiring multiple GPUs for large datasets and high memory consumption.
Built on Scanpy and rapids-singlecell, ScaleSC represents a major leap forward in single cell data analysis. ScaleSC enables significantly faster processing of single cell datasets compared to Scanpy. As shown in Table 1, for a medium-scale dataset of 1.3 million mouse brain cells, ScaleSC completed the task in 2 minutes, whereas Scanpy required 4.5 hours to process the entire dataset. Additionally, ScaleSC can process 13 million cells within an hour, surpassing rapids-singlecell’s limit of 1 million cells without multi-GPU support. These benchmarks were conducted on a system running Rocky Linux 8.10, equipped with 1 TB of CPU RAM and a single NVIDIA A100 GPU.

Table 1. The time cost comparison between Scanpy and ScaleSC on the 1.3M mouse brain data.
How does ScaleSC accomplish this? It overcomes memory bottlenecks through smart chunking strategies. However, chunking introduces new challenges in data calculation and final result generation. New algorithms were developed for cell QC, highly variable gene (HVG) detection, PCA, and batch correction. Fig. 1 illustrates the chunk-based data loading and preprocessing workflow.
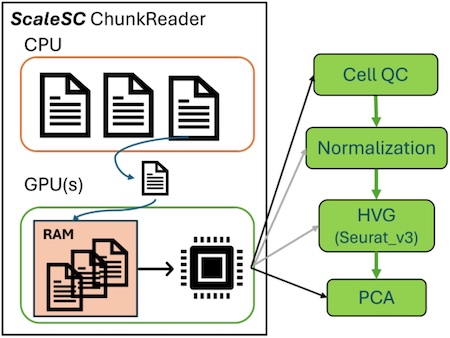
Figure 1. The flowchart Chunk-based data loading and preprocessing.
Beyond computational efficiency, accuracy is also a crucial consideration. Scanpy and rapids-singlecell implement the same algorithms slightly differently in order to optimize the different computational architectures between CPU and GPU. These differences result in computational inconsistencies. ScaleSC introduces several key innovations and resolves discrepancies in the calculations of PCA, K-means, and nearest neighbor analysis. As a result, ScaleSC can replicate Scanpy’s results across most analysis steps.
Cell type annotation is essential for cell type decomposition analysis and understanding biological processes, such as pathways enriched in certain cell types. Cell type annotation follows two main approaches: de novo annotation, which relies on marker genes, and reference mapping, which transfers annotations from well-annotated datasets.
By leveraging modern GPU’s parallel computing capabilities, ScaleSC integrates NSForest (Liu et al.,2024) for marker gene identification in De novo cell type annotation. This approach enables the analysis of multi-millions-cell datasets while maintaining NSForest’s strong feature selection performance. For a medium-scale scRNA-seq dataset containing 661, 789 cells and 36, 341 genes, ScaleSC’s results were compared to those obtained using Scanpy. As shown in Fig. 2, the marker genes identified by ScaleSC demonstrated superior biological relevance, characterized by both increased cell-type specificity and elevated expression levels.
.jpg)
Figure 2. Expression of gene markers between Scanpy and ScaleSC is compared using a dataset with 0.6M cells.
Looking Forward:
With its speed, scalability, and accessibility, ScaleSC is poised to become an essential tool for single cell analysis. For researchers analyzing large-scale single cell data, ScaleSC offers a powerful new solution that doesn't require access to extensive computational resources. By accelerating discoveries in single cell genomics, it has the potential to uncover new insights into cellular biology and disease.
Freely available on GitHub (https://github.com/interactivereport/ScaleSC), ScaleSC empowers researchers worldwide to leverage its capabilities. BioInfoRx also plans to integrate ScaleSC into the BxGenomics workflow.
References:
1. Hu, W., Zhang, H., Sun, Y. H., Cao, S., Gagnon, J., Ouyang, Z., Moroishi, Y., & Zhang, B. (2025). ScaleSC: A superfast and scalable single cell RNA-seq data analysis pipeline powered by GPU. bioRxiv. https://doi.org/10.1101/2025.01.28.635256
2. Wolf, F. A., Angerer, P., & Theis, F. J. (2018). Scanpy: Large-scale single-cell gene expression data analysis. Genome Biology, 19, 1–5. https://genomebiology.biomedcentral.com/articles/10.1186/s13059-017-1382-0
3. Dicks, S., Angerer, P., Pintar, J., Korten, T., Kumar, A., Virshup, I., & Metzger, P. (2024). scverse/rapids singlecell: v0.10.6. Zenodo. https://zenodo.org/records/12533399
4. Liu, A., Peng, B., Pankajam, A. V., Duong, T. E., Pryhuber, G., Scheuermann, R. H., & Zhang, Y. (2024). Discovery of optimal cell type classification marker genes from single-cell RNA sequencing data. BMC Methods, 1(1), 15. https://pubmed.ncbi.nlm.nih.gov/38712147/